Prediction of chemical reaction yields using deep learning
The research article "Prediction of chemical reaction yields using deep learning" was recently published in Machine Learning: Science and Technology.
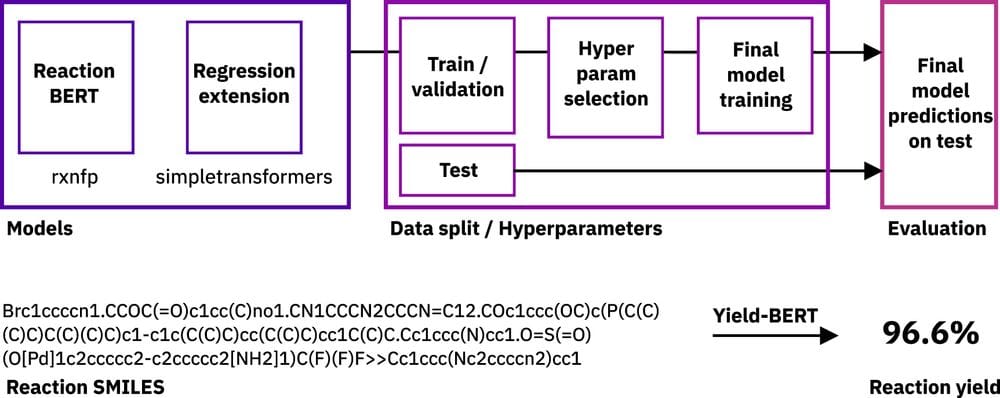
Artificial intelligence is driving one of the most important revolutions in organic chemistry. Multiple platforms, including tools for reaction prediction and synthesis planning based on machine learning, have successfully become part of the organic chemists' daily laboratory, assisting in domain-specific synthetic problems. Unlike reaction prediction and retrosynthetic models, the prediction of reaction yields has received less attention in spite of the enormous potential of accurately predicting reaction conversion rates. Reaction yields models, describing the percentage of the reactants converted to the desired products, could guide chemists and help them select high-yielding reactions and score synthesis routes, reducing the number of attempts. So far, yield predictions have been predominantly performed for high-throughput experiments using a categorical (one-hot) encoding of reactants, concatenated molecular fingerprints, or computed chemical descriptors. Here, we extend the application of natural language processing architectures to predict reaction properties given a text-based representation of the reaction, using an encoder transformer model combined with a regression layer. We demonstrate outstanding prediction performance on two high-throughput experiment reactions sets. An analysis of the yields reported in the open-source USPTO data set shows that their distribution differs depending on the mass scale, limiting the data set applicability in reaction yields predictions.
Author(s): Philippe Schwaller, Alain C. Vaucher, Teodoro Laino and Jean-Louis Reymond