The Chemical Space Project
Introduction
In chemistry one spends a lot of time designing and making molecules. These molecules should be new, interesting, and do what they are supposed to do. To succeed in this game, you need to know at least what’s already known, which for organic molecules corresponds to over 100 million molecules and thousands of different methods to make them. These very large numbers have led to the concept of chemical space, which is a space where molecules are distributed according to their structures and properties, making it easier for the human mind to grasp their diversity.
In our group, we develop computational tools to enumerate, visualize and search chemical space, mostly to identify new drugs. We use these methods to design small molecules and peptides, which we synthesize and test in the laboratory. Our group is interdisciplinary and touches on computer science, cheminformatics, organic synthesis, medicinal chemistry, peptide synthesis, microbiology and structural biology. Projects include small molecules targeting ion channels and transporters (NCCR TransCure), peptide-based antibiotics (NRP72 antimicrobial resistance), DNA/siRNA transfection (MMBio), anti-inflammatory and cell-targeting reagents. We also design and synthesize complex carbohydrates to study protein glycosylation enzymes (GlycoSTART).
GDB
It’s quite difficult to design a new molecule. Things would be a lot easier if a list of all the possible molecules would be available to choose from. The Generated DataBase GDB17 is such a list for small organic molecules up to 17 atoms (which may be C, N, O, S, or halogen, not counting hydrogen atoms). This database contains 166.4 billion molecular structures. We also have smaller subsets focusing on molecules for drug discovery, such as the fragment database FDB17, the medicinal chemistry aware database GDBMedChem, and the ChEMBL-like database GDBChEMBL.
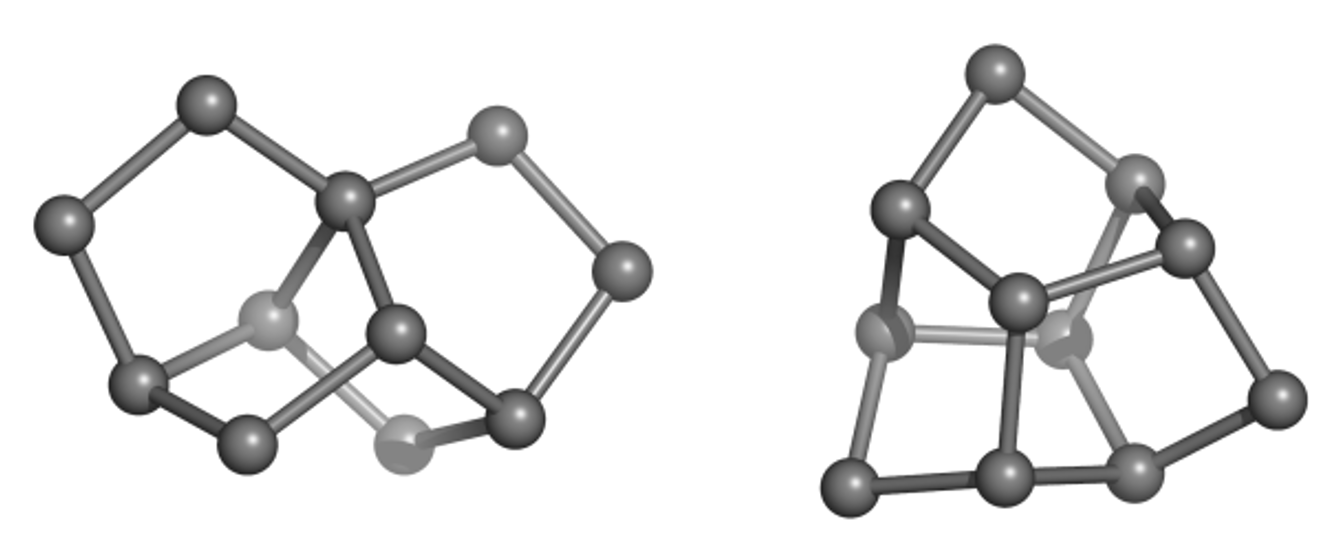
Much of the originality and novelty of GDB molecules lies in new combinations of rings, called ring systems. The database GDB4c lists all possible ring systems up to 4 rings and a maximum of 30 atoms (not counting hydrogen atoms). Like all our GDBs, GDB4c is a treasure trove of new molecules because the vast majority of GDB molecules (>99%) are novel. Examples of unusual but accessible GDB ring systems include trinorbornane (Figure 1), which we showed as a possible molecule in 2007 and was synthesized in 2017 by the group of Marcel Mayor in Basel. The tris-homocubane at right, which also contains three norbornane units in an unusual chiral arrangement, is sofar only known as the tri-oxa variant obtained by acid-catalyzed cyclization of the C3-symmetrical tri-epoxide of barrellene.
Peptides
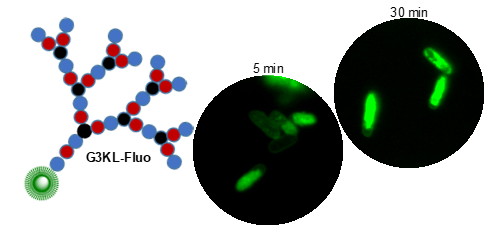
Peptides are chains of amino acids linked by amide bonds. These chains are flexible and, depending on the sequence of amino acids, may fold and cyclize into organized 3D-structures or self-assemble into supramolecular aggregates. Many peptides are biologically active (hormones, antibiotics, toxins). To expand the chemical space of natural peptides, which are mostly linear or cyclic, we have developed the synthesis of peptides containing branching points to obtain other chain topologies. We focus on peptide dendrimers, which are molecular trees made of amino acids, and on bicyclic peptides. We have discovered antimicrobial peptides (AMPs) selectively disrupting bacterial membranes and which we are developing for therapeutic use against multidrug resistant bacteria (e.g. G3KL, Figure 2). Further projects include peptide dendrimers for DNA and siRNA transfection and as anti-inflammatory and cell-targeting agents.
Our initial discoveries of peptide dendrimers were driven by chemical synthesis and activity screening. More recently we have established computational tools to navigate the peptide chemical space and improve both discovery and optimization. Our most recent such tool is the peptide design genetic algorithm PDGA, which allows us to explore the chemical space of peptides of any chain topology (linear, cyclic, polycyclic, dendritic) despite of its overwhelming size and complexity (over 1030 possible peptides).
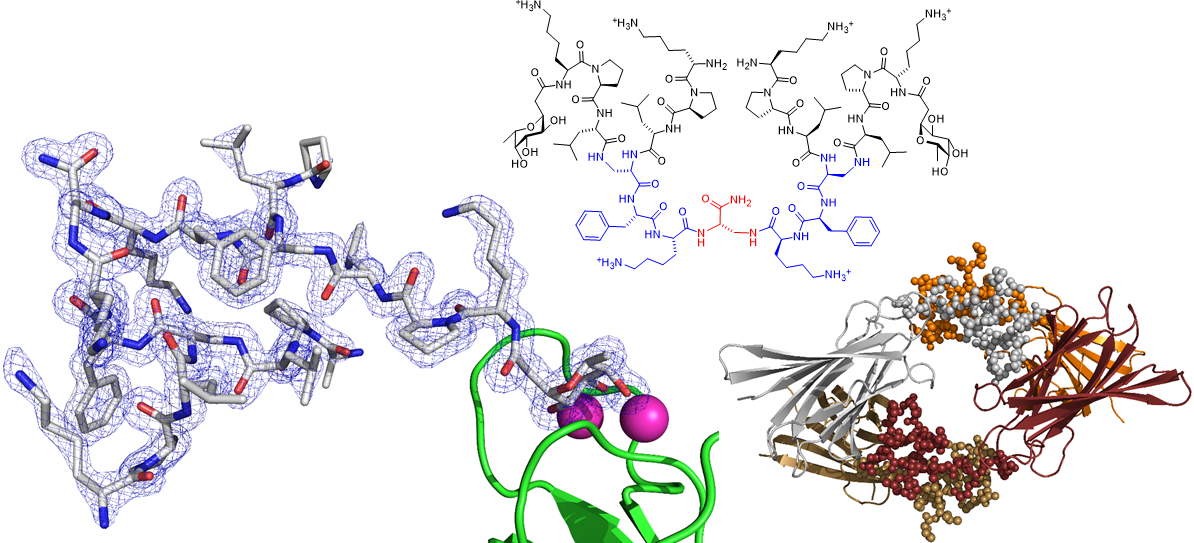
We also spend a lot of time investigating the structure and properties of our peptides. We use x-ray crystallography of glycosylated analogs as complexes with the microbial lectin LecB as well as molecular dynamics simulations. Recent successes with this approach include the first X-ray crystal structure of short α-helical linear AMPs, and the first X-ray crystal structure of a peptide dendrimer, which revealed how intramolecular contact between branches stabilize its secondary structure (Figure 3).
Chemical Space Maps
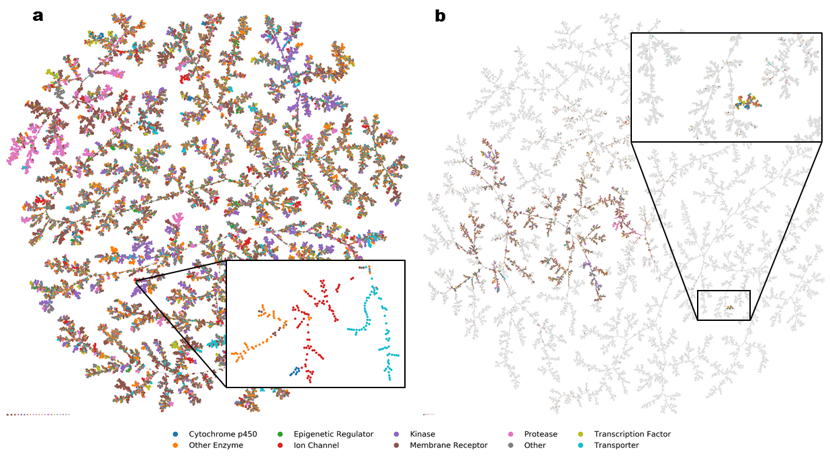
Starting from a database of molecules, we map its chemical space in two steps. First, we compute for each molecule a set of N numbers, called fingerprint, which encodes its molecular structure and defines a point in an N-dimensional space. Our favourite fingerprints are MQN (Molecular Quantum Numbers, 42 indices counting atoms, bonds, polar groups and topological features), MHFP6 (MinHash fingerprint, representing circular substructures) and MXFP (Macromolecule extended atom-pair fingerprint, 217 counts of atom pairs representing molecular shape). Second, we project the N-dimensional space into 3D or 2D to obtain an interactive map, which features color-coded points corresponding to molecules, and groups molecules according to their structural similarities. Map formats include mapplets, web-based 3D-maps (faerun), virtual reality, and most recently TMAP (tree-map), a very powerful layout to inspect large high-dimensional datasets not only in chemistry but also in other areas of science and culture (Figure 4).
Drug Discovery
The fingerprints used to map chemical space can be used in drug discovery for hit finding (sampling), hit or lead optimization (nearest neighbour searches), and to assess polypharmacology (target prediction and identification). Various online tools such as PPB2 (Figure 5) that were developed in our group are available on the gdb webpage.
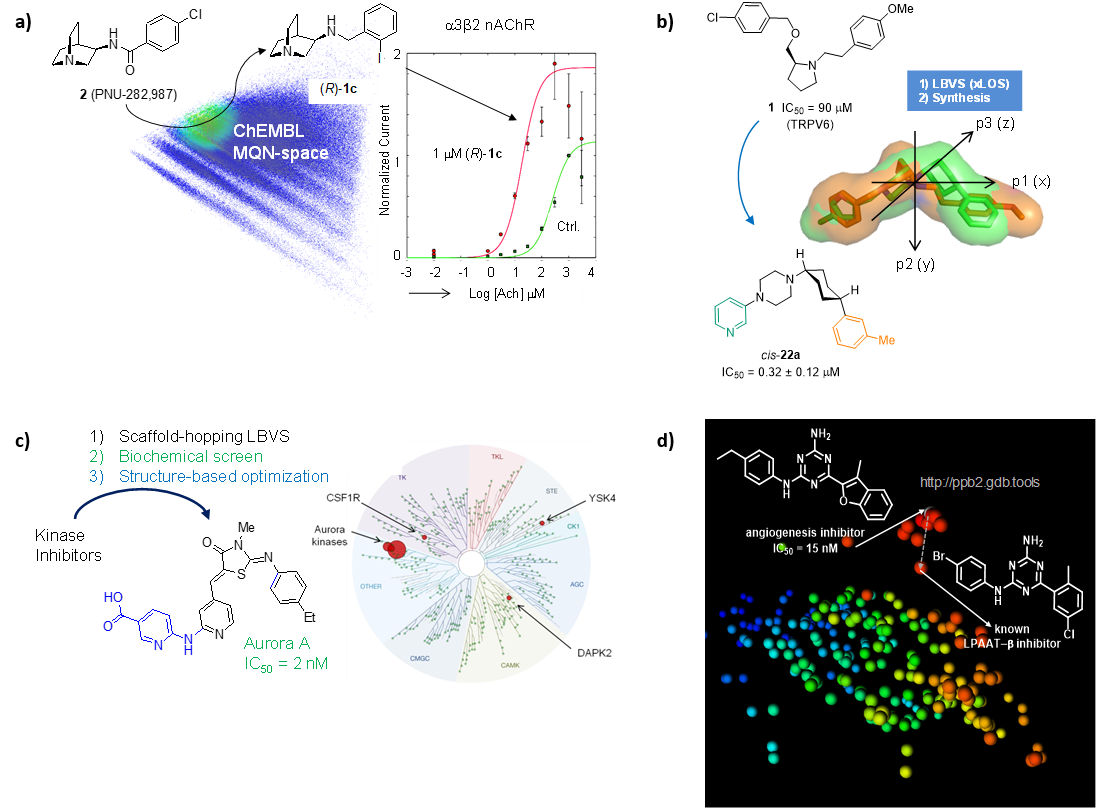
We have used similarity searches in chemical space to identify and optimize small molecules targeting ion channels (NDMA receptors, α7/α3β2 nAChR, TRPV6, TRPM4) and transporters (GLT-1, DMT1). An example spanning hit finding, lead optimization, off-target profiling and determination of target binding mode by X-ray crystallography is the discovery of a highly selective inhibitor of the kinase Aurora A, an important cancer target. We have also used nearest neighbour searches to identify the target of a hit compound from an angiogenesis inhibition phenotypic screen (LPAAT-β) (Figure 6).
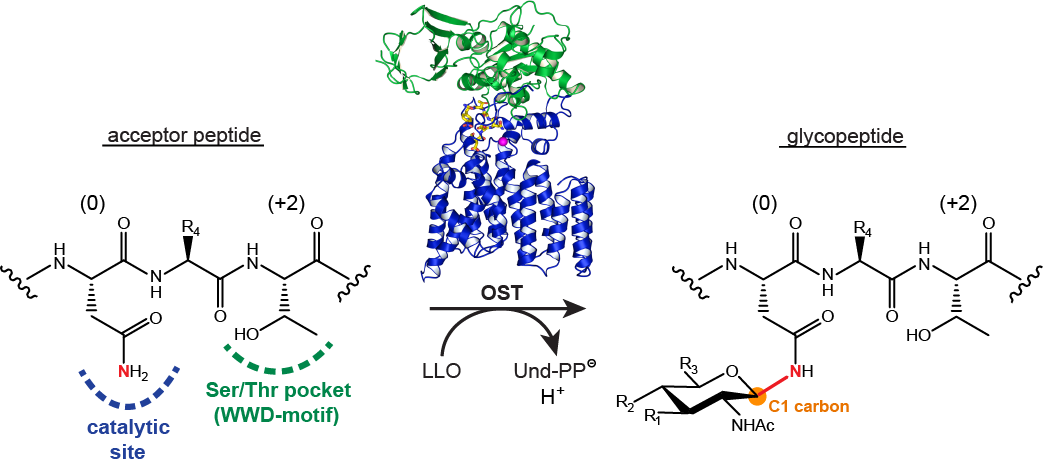
Protein Glycosylation
Following on previous activities in developing reagents for enzyme assays and glycosidase inhibitors, we collaborate with Markus Aebi and Kaspar Locher at ETHZ to study protein glycosylation enzymes, in particular oligosaccharyltransferases (Figure 7) and oligosaccharide flippases. In this project we design and synthesize fluorescent peptide and lipid-linked oligosaccharide model substrates to enable biochemical and structural studies.