Deep Learning Invades Drug Design and Synthesis
The commentary Deep Learning Invades Drug Design and Synthesis has been published by Chimia.
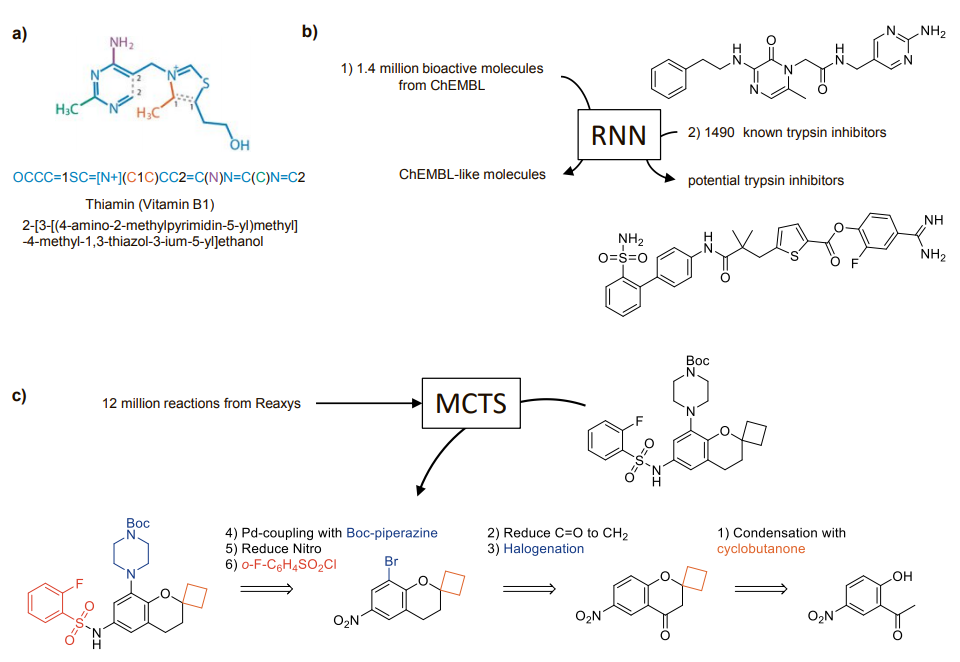
Discovering a new drug goes asfollows: given a medical need, identify the underlying biological mechanism, and design a molecule acting via this mechanism to produce the desired effects. This sounds simple, but in truth it’s not easy at all. Leaving aside biology, an important part of the problem is the chemistry: there are just too many molecules to choose from, perhaps as many as 1060 for all drug-like small molecules. Even with the help of computers one cannot enumerate more than a few billions of them, let alone predict their possible biological activity or how to synthesize them. Of course, we don’t really need to look at all these molecules. We can rely on trained medicinal chemists to make educated guesses on which onesto make and test first, often aided by modeling and automated searches, and this works well enough that trial-and-error cycles eventually succeed. Here comes a disruptive idea: can we automate the process and take the chemist out of the equation? Recent papers suggest that this might become possible using deep learning. Deep learning is an umbrella term for machine learning methods based on artificial neural networks, by which a software first learns from training data, and then is able to perform complex tasks or predictions. Parallelization of computer calculations, graphic cards and software frameworks such as TensorFlow have made deep learning practical for writing music, translating languages, and even playing (and winning) Go.
Author(s): Josep Arús-Pous, Daniel Probst, and Jean-Louis Reymond